MT-DTI, 세계 최고 성능의 ‘약물-단백질 상호작용 예측’ 모델
디어젠(주)는 세계 최고 수준의 약물-단백질 상호작용 예측 모델 MT-DTI 대해서 발표했습니다. 연구 결과는 JMLR(Journal of Machine Learning Research)저널에 게재되어 있습니다.

Introduction
약물-단백질 상호작용(Drug Target Interaction, DTI)은 신약발견 프로세스에서 핵심적인 역할을 합니다. 질병의 원인이 되는 비정상적인 수치의 단백질을 조절해주는 약물 후보군을 방대한 약물 후보군으로부터 추려주는 역할을 하기 때문입니다. 전통적으로는 실험실 기반의(in-vitro) DTI를 시행해 왔기 때문에 비용과 시간이 많이 소요됐지만, 최근에는 컴퓨터 시물레이션 기반의(in-silico) DTI 모델이 등장하여 효과적인 신약발견 프로세스를 위한 시도가 이루어지고 있습니다.
이러한 시도로 머신러닝과 딥러닝에 기반한 다양한 기법이 제안되었지만, 이들은 복잡한 화학구조를 제대로 반영하지 못하고 방대한 화합물 정보를 제대로 이용하지 못하여 후보물질예측에 한계가 있는 것으로 알려져 있습니다. 이러한 문제를 해결하기 위해서 이 논문에서는 Self-Attention 메커니즘을 DTI 모델에 반영해서 복잡한 화학구조를 더욱 효과적으로 모델링 했고 PubChem의 약 9700만 개의 화합물 데이터베이스를 사전학습에 이용해서 모델의 정밀도를 높였습니다.
Background

DTI 문제는 (약물, 단백질) 쌍을 입력으로 받아서 두 입력의 binding 정도를 뜻하는 score를 예측하는 문제입니다. (그림 1) 약물과 단백질 모두 sequence 형태로 들어오게 되는데 약물은 SMILES라고 불리는 sequence로 원자와 결합정보를 담고 있고 단백질은 FASTA라고 불리는 sequence로 여러 아미노산의 배열로 구성되어 있습니다. 이렇게 문제를 형식화하면 DTI를 머신러닝의 regression 문제로 일반화 할 수 있어서 여러 가지 모델을 시험해 볼 수 있습니다. 일례로, 본 논문 이전의 가장 최신 DTI 모델은 DeepDTA인데 2D convolution 연산에 기반한 모델입니다.
Motivation

Convolution은 squence 상에서 인접한 token들과 함께 연산이 되는 특징이 있습니다. 하지만 항상 유용한 정보가 인접한 token에 있으리라는 보장은 없습니다. 예를 들어, 유명항암제의 3차원 구조와 sequence 표기법을 나타낸 그림 2를 보면, Cl과 F는 sequence 상으로 인접해 있어서 연산이 같이 되지만 이들보다 실제 거리가 가까운 Cl과 N의 경우에는 sequence 상으로는 멀리 떨어져 있어서 연산이 이루어지지 않습니다. 그래서 정확한 약물 모델링에 한계가 있을 수밖에 없습니다.
또한, 기존 DTI 모델들은 방대한 저분자화합물 데이터베이스를 이용할 수 없는 구조적 문제가 있습니다. 따라서 본 논문에서는 Self-Attention 메커니즘을 모델에 반영해서 모든 토큰을 효과적으로 고려할 수 있도록 했을 뿐 아니라 기존 저분자화합물 데이터베이스로부터 화합물 합성패턴에 대한 지식을 효율적으로 추출하여 모델에 반영했습니다.
Method

저분자 화합물을 효과적으로 모델링 하기 위해서 우리는 Molecule Transformer (MT, 그림 3) 를 제안했습니다. 이는 Self-Attention을 기반으로 한 encoder들을 여러 겹 쌓아 올린 형태로 구성되어 있습니다. MT가 저분자 화합물에 대한 규칙과 같은 핵심 지식을 학습할 수 있도록 PubChem의 약 9700만 개의 화합물에 대해서 pre-train(사전학습)을 수행했습니다. 그 결과 SMILE 시퀀스의 문법예측 태스크를 97% 이상의 정확도로 수행할 수 있었습니다.
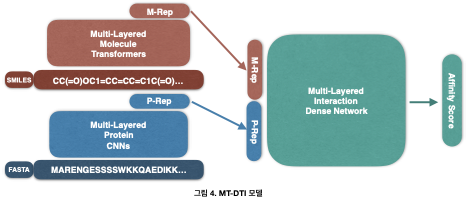
이렇게 학습된 MT를 그림 4의 DTI 모델에 그대로 삽입하여 DTI 데이터셋에 대해서 fine-tuning (정밀학습)을 수행합니다. 이렇게 모델의 한 부분을 pre-train 후에 큰 모델에 삽입하여 fine-tuning 하는 작업을 transfer learning (전이 학습)이라고 부르는데 원하는 작업과 관련된 데이터가 부족한 경우에 아주 유용합니다. 왜냐하면 pre-train 된 모델은 원하는 작업의 데이터는 아니지만, 그 데이터와 유사한 방대한 데이터에서 중요한 정보들을 습득하기 때문입니다.
Experiments

실험은 두 가지 데이터에 대한 네 가지 성능 측도에 대한 비교를 수행했습니다. Kiba와 Davis 모두 DTI 데이터로써 (약물, 단백질)에 대한 각각의 기준이 되는 바인딩 정도를 나타내는 점수로 구성되어있습니다. CI (concordance index)와 r^2_m 값은 regression에 대한 성공 척도이고, MSE는 모델의 예측과 실제 값과의 차이를 나타내며, AUPR은 regression을 binary 문제로 치환하여 binding 여부를 예측했을 때의 정확도를 나타냅니다.
본 논문에서 제시한 모델은 MT-DTI이고, w/o FT이 붙은 모델은 fine-tuning 작업을 생략한 MT-DTI 모델입니다. 비교에 쓰인 모델은 최신 머신러닝과 딥러닝 기반의 모델들을 사용했습니다. 실험 결과 MT-DTI가 기존 방법들과 비교했을 때 모든 성능 측도에서 우수한 성능을 보였습니다. 또한 fine-tuning을 생략했을 때에도 기존 방법들과 비슷하거나 때로는 더 잘 나오는 경우도 있었습니다. 이는 self-attention 기반의 모델링이 복잡한 화합물을 효과적으로 표현할 수 있었기 때문입니다.
Case Study

이전 실험 결과에서는 제안된 MT-DTI 모델은 binding score를 정확하게 예측할 수 있음을 실험에서 보였습니다. 하지만 실제로 이 모델이 얼마나 신약개발 과정에서 의미 있게 사용될 수 있을지를 추가 실험을 통해 살펴보았습니다.
Drugbank라는 FDA에서 승인된 1,794개의 약물들과 EGFR이라는 유명한 암 바이오마커 단백질을 함께 제안된 모델에 입력으로 넣고 30개의 EGFR 타깃 약물 후보군을 추려보는 실험을 했습니다. 그 결과 놀랍게도 Drugbank에 존재하는 실제 EGFR 타깃 약물 8개가 모두 후보 중에 있었음을 발견했습니다. 이는 제안된 모델의 유용성을 증명했을 뿐 아니라 다른 22개의 제안된 약물 후보들도 잠재적 항암제로 검증해 볼 필요가 있다는 뜻입니다.
Conclusion
제안된 MT-DTI 모델은 기존 DTI 모델들이 갖고있는 한계점에 착안하여 최신 딥러닝 기술을 효과적으로 도입한 모델로서 두 가지 표준화된 DTI 데이터에서 가장 좋은 성능을 보였습니다. 또한 실제 case study를 통해서 제안된 모델이 신약발견 과정에서 효과적으로 이용될 수 있음을 보였습니다. 하지만 self-attention 기법은 DTI의 입력중 하나인 약물쪽에만 적용되었는데 이를 단백질에도 확장을 해본다면 더 좋은 모델을 기대할 수 있을 것입니다.
이를 위해서는 많은 양의 단백질 데이터셋을 확보해야 합니다. 또한 self-attention은 짧은 sequence에만 적용이 용이하기 때문에 긴 sequence를 효과적으로 다룰 수 있는 새로운 모델을 개발한다면 더 좋은 DTI 모델을 기대해 볼 수 있을 것입니다.
Test Our MT-DTI
MT-DTI모델은 아래 버튼을 통해 사용해 볼 수 있습니다. 자신만의 데이터를 이용하여, 손쉽게 약물-단백질 상호작용 score를 확인해 보세요.